- Home
- Genomes
- Genome Browser
- Tools
- Mirrors
- Downloads
- My Data
- Projects
- Help
- About Us
WHAT IS A WOBBLE BASE PAIR?
A wobble base pair is a base pair between an mRNA and the anticodon of a tRNA that does not follow the "normal" Watson-Crick base pairing where adenine pairs with thymine — or uracil in RNA — and guanine pairs with cytosine. An amino acid is typically encoded by a codon sequence in the mRNA (as seen in Figure 1), but with wobble base pairing, the pairing between the third nucleotide in the codon and first nucleotide in the anticodon of the tRNA that carries the new amino acid to the nascent peptide chain may not be a Watson-Crick base pair. This allows a single tRNA to serve more than one codon and yet add the same amino acid to a protein during synthesis.
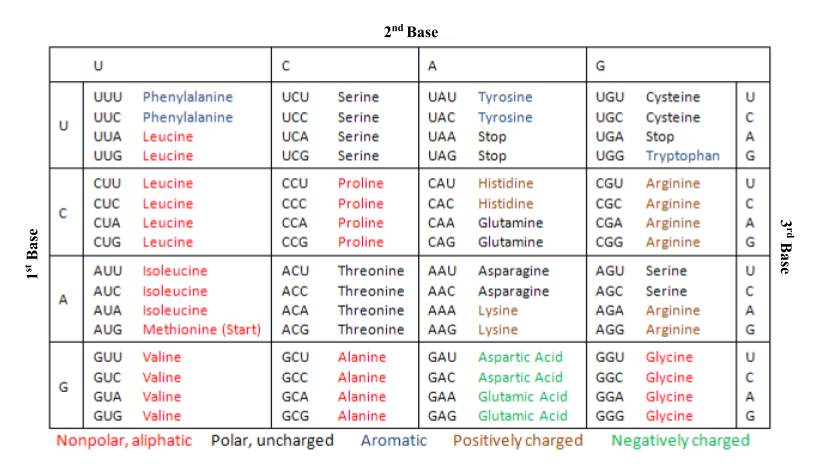
Figure 1.Codon table representing the amino acids coded by each mRNA codon. As seen for alanine, leucine, and several other amino acids, an amino acid can be encoded by several codons. — modified from wikipedia.
WOBBLE BASE PAIRS IN THE GENOME BROWSER
Using various features in the UCSC Genome Browser, we have the ability to visualize changes through evolutionary time, in homologous codons where the third base varies and yet the same amino acid appears through various species.
Figure 2 is a representation of chr4:3,037,158-3,037,180 on human assembly hg19 with the base position, UCSC genes, PhyloP conservation Score, and Conservation 100-way tracks displayed. As seen in the figure, each codon sequence of three nitrogenous bases encodes its respective amino acid listed in the codon table in figure 1. GCG produces an alanine, GCT produces another alanine, GGG produces a glycine, etc. The track following UCSC genes is the "100 Vertebrates Basewise Conservation by PhyloP" track,
which displays the conservation of the bases across species.
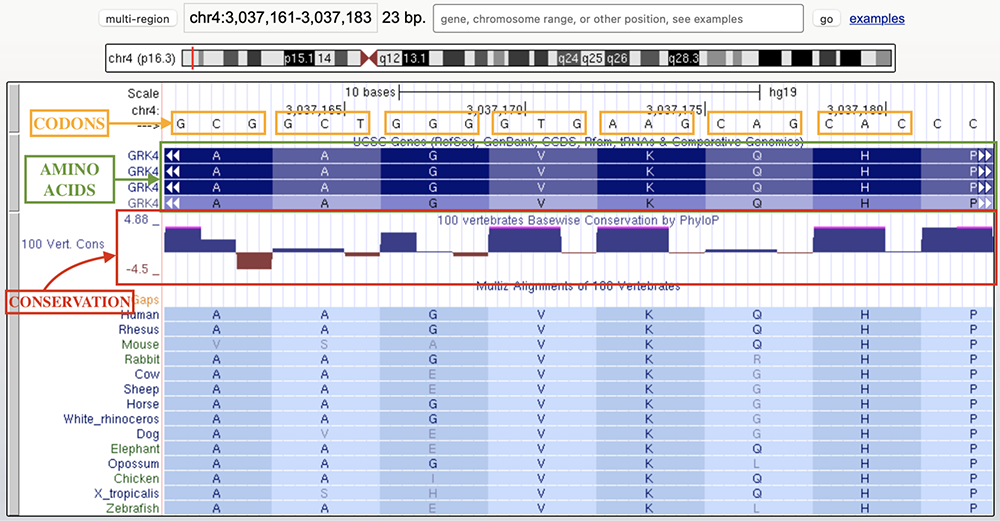
Figure 2. Genome session portraying wobble bases. For the valine (V) and lysine (K) residues in the center of the figure, the third base is variable through evolution but the amino acid is invariant.
In figure 3, the conservation score of a codon sequence encoding a valine amino acid is shown. The first two bases (G and T) have a score above the 4.88 cutoff value in the figure (shown as a pink bar on top of the peak), while the third base (G in human) has a score of -0.0323386; this means that while the first two bases are highly conserved, the last is not. However, despite the low conservation of the third base, the amino acid across different species does not change and is still a valine. In this example, the change of the third nucleotide on an evolutionary level can still make the same amino acid (hence, a wobble base), but this is not always the case.
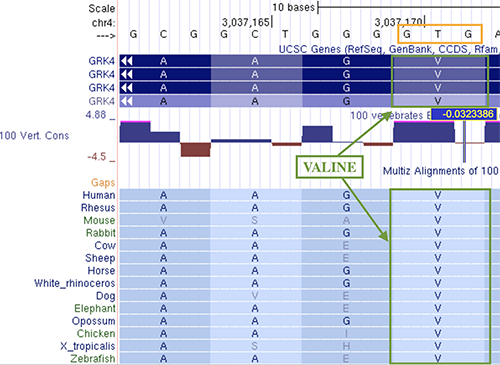
Figure 3. Conservation score of codon sequence for amino acids.
As seen in figure 4, in both the first and second codon sequence encoding alanines, the first and second bases are conserved while the third base is not. The amino acids produced in most species are alanine; however, some have a change in its amino acids, such as in mice. The first codon results in valine while the second codon results in a serine instead of alanine.
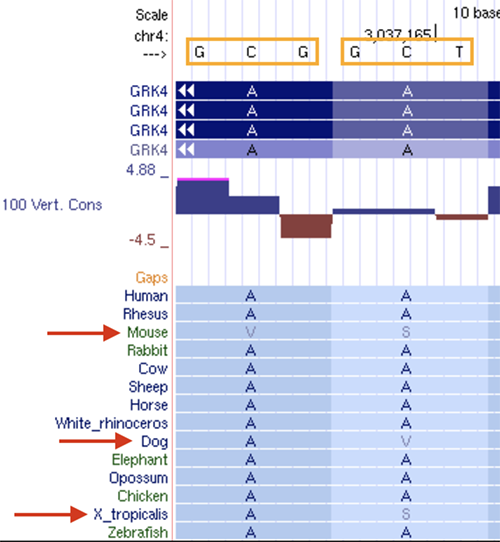
Figure 4. Changes in amino acid due to low conservation in the second base of the codon.
AA > VS in mouse; AA > AV in dog; AA > AS in frog (X. tropicalis)
To further view wobble bases in the genome browser, we can turn on the Pfam track and include a few more species from each
clade —
 https://genome.ucsc.edu/s/education/hg19_wobble2
The Pfam track displays conserved regions across species and across proteins in the same species; oftentimes, these regions are functionally important parts of the protein. They therefore have certain amino acids that are very well conserved through many species, indicating a functional role for those amino acid residues, such as the protein kinase domain in the GRK4 gene shown in the figure. Note the high conservation score for the first two nucleotides of the codons for valine, lysine, histidine and proline residues in the session (but not the glutamine): (VKqHP).
https://genome.ucsc.edu/s/education/hg19_wobble2
The Pfam track displays conserved regions across species and across proteins in the same species; oftentimes, these regions are functionally important parts of the protein. They therefore have certain amino acids that are very well conserved through many species, indicating a functional role for those amino acid residues, such as the protein kinase domain in the GRK4 gene shown in the figure. Note the high conservation score for the first two nucleotides of the codons for valine, lysine, histidine and proline residues in the session (but not the glutamine): (VKqHP).
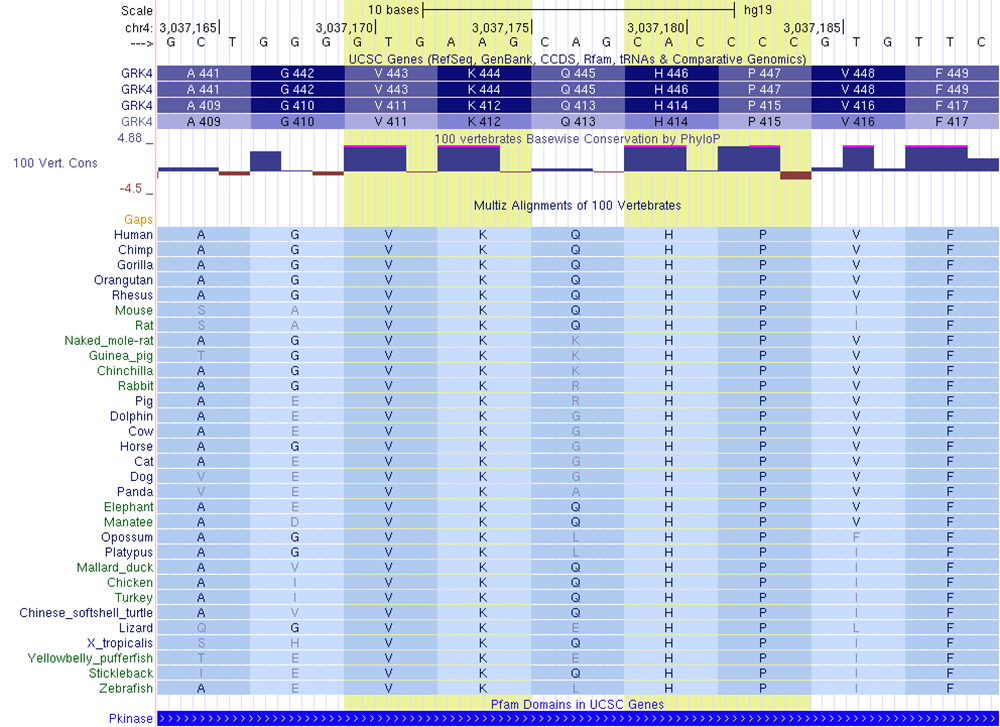
Figure 5. Highly conserved region showing four codons out of five with very high PhyloP scores. PFAM track shows that this is a functionally significant protein kinase domain. Conservation implies functional significance.
Written by Callie Lin, UCSC. Majors: BS, Biomolecular Engineering & Bioinformatics; BS, Neuroscience