new
Note: September 30, 2024
Description
The Genome Aggregation Database (gnomAD) - Rare CNV variants (<1% overall site frequency) v4.1 track set shows rare autosomal coding copy number variants (CNVs) with an overall
site frequency of less than 1%. These variants were identified from exome sequencing (ES) data of
464,297 individuals. The data can also be explored via the
gnomAD browser.
Display Conventions and Configuration
Items are colored by the type of variant:
| Variant Type |
| Deletion (DEL) |
20989 |
| Duplication (DUP) |
25026 |
.
Mouseover on an item will display the position, size of variant, genes impacted by
variant (>=10% CDS overlap by deletion or >=75% CDS overlap by duplication), and site
frequency of non-neuro control samples. Item description pages include a linkout to
the gnomAD browser showing additional genetic ancestry group information.
Methods
Exome CNV Discovery Method: GATK-gCNV
To identify rare coding CNVs from the ES data of 464,297 individuals in gnomAD v4, the GATK-gCNV
method was employed, as described in Babadi et al., Nat Genet, 2023.
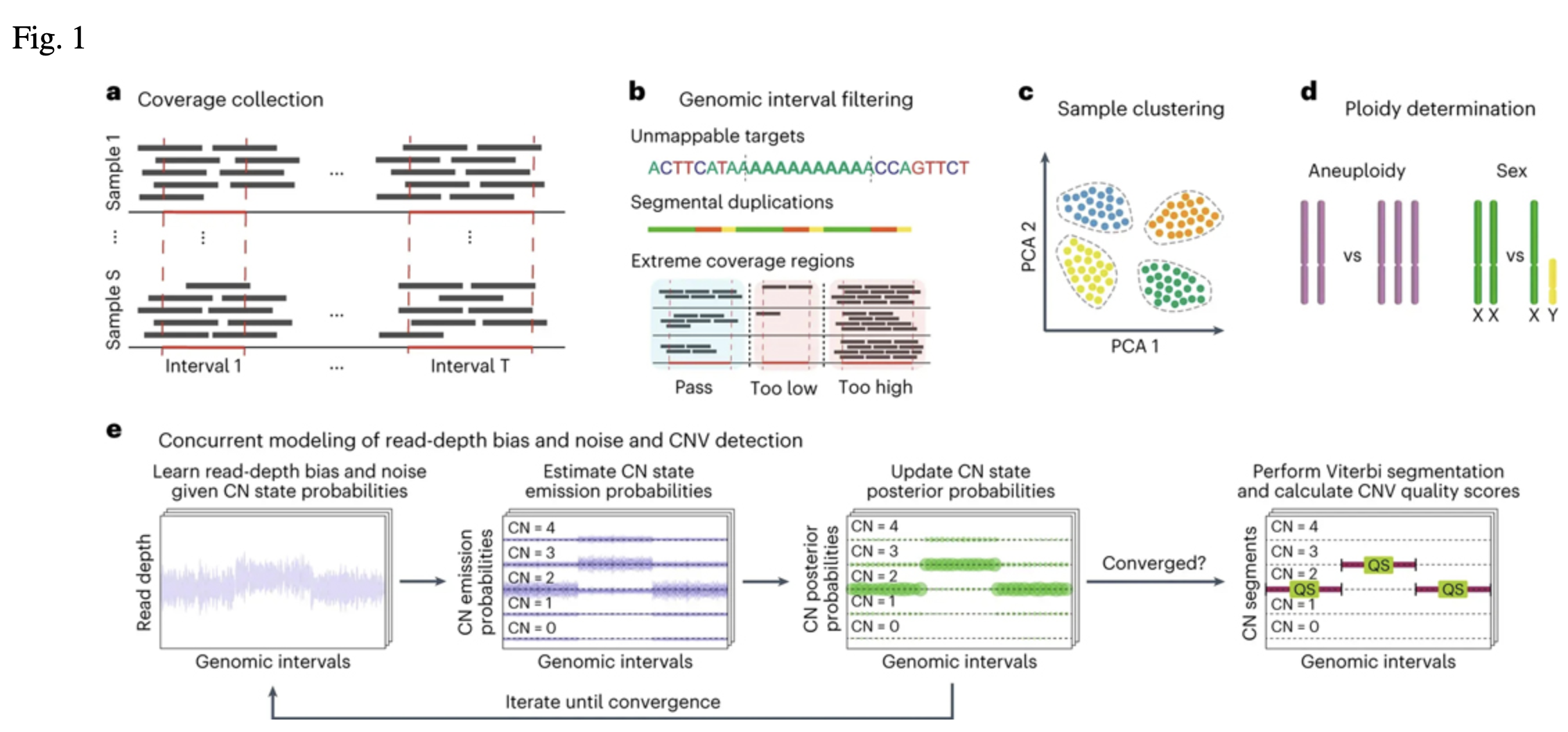
The CNV discovery process started with collecting the number of reads mapped to 363,301 autosomal
target intervals derived from protein-coding exons (Fig. 1a, b; Babadi et al.). These read counts
were used to capture sample-level technical variability, such as differences in exome capture kits
or sequencing centers, and generated 1,045 different batches of samples for parallel processing
(Fig. 1c). For each of these batches, 200 random samples were selected for training GATK-gCNV in
cohort mode,which can be thought of as the creation of a "panel of normals" (PoN). The resulting
PoN models were then used to efficiently delineate CNV events on all of the samples of their
respective cohorts using the GATK-gCNV case mode (Fig. 1d,e).
The raw, individual-level CNV calls produced by GATK-gCNV for all samples were then collated,
and variants observed in multiple individuals were clustered using single-linkage clustering.
Quality filtering followed the procedures outlined in Babadi et al., filtering CNVs based on
sample-level (number of events per individual) and call-level (frequency, size, quality score) metrics
Due to the significant increase in cohort size and heterogeneity compared to the datasets reported
in Babadi et al., additional filters were applied. Samples with more than five chromosomes harboring
rare CNVs, as well as those containing more than three rare terminal CNVs, were excluded. 1,049
sites producing noisy normalized read-depth signals were masked. The final retained CNVs and sites
were subsequently annotated for impacted genes and frequencies.
Limitations of ES-based rare coding CNVs in gnomAD v4
- This dataset includes only rare coding CNVs, filtered to <1% site frequency in the overall
dataset.
- This dataset only includes variants that span three or more exons that received sufficient
coverage.
- This dataset is limited to autosomal CNVs for now.
More information can be found at the
gnomAD site.
The bed files was obtained from the gnomAD Google Storage bucket:
https://storage.googleapis.com/gcp-public-data--gnomad/release/4.1/exome_cnv/gnomad.v4.1.cnv.non_neuro_controls.bed
The data was then transformed into a bigBed track. For the full list of commands used to make this
track please see the "gnomAD CNVs v4.1" section of the
makedoc.
Data Access
The raw data can be explored interactively with the Table Browser, or
the Data Integrator. For automated access, this track, like all
others, is available via our API. However, for bulk
processing, it is recommended to download the dataset. The genome annotation is stored in a bigBed
file that can be downloaded from the
download server.
The exact filenames can be found in the track configuration file. Annotations can be converted to
ASCII text by our tool bigBedToBed which can be compiled from the source code or
downloaded as a precompiled binary for your system. Instructions for downloading source code and
binaries can be found
here. The tool can
also be used to obtain only features within a given range, for example:
bigBedToBed http://hgdownload.soe.ucsc.edu/gbdb/hg38/gnomAD/v4/cnv/gnomad.v4.1.cnv.non_neuro_controls.bb -chrom=chr6 -start=0 -end=1000000 stdout
Please refer to our
mailing list archives
for questions and example queries, or our
Data Access FAQ
for more information.
More information about using and understanding the gnomAD data can be found in the
gnomAD FAQ site.
Credits
Thanks to the Genome Aggregation
Database Consortium for making these data available. The data are released under the ODC Open Database License
(ODbL) as described here.
References
Babadi M, Fu JM, Lee SK, Smirnov AN, Gauthier LD, Walker M, Benjamin DI, Zhao X, Karczewski KJ, Wong
I et al.
GATK-gCNV enables the discovery of rare copy number variants from exome sequencing data.
Nat Genet. 2023 Sep;55(9):1589-1597.
PMID: 37604963; PMC: PMC10904014
Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C,
Gauthier LD, Wang H et al.
A structural variation reference for medical and population genetics.
Nature. 2020 May;581(7809):444-451.
PMID: 32461652; PMC: PMC7334194
Cummings BB, Karczewski KJ, Kosmicki JA, Seaby EG, Watts NA, Singer-Berk M, Mudge JM, Karjalainen J,
Satterstrom FK, O'Donnell-Luria AH et al.
Transcript expression-aware annotation improves rare variant interpretation.
Nature. 2020 May;581(7809):452-458.
PMID: 32461655; PMC: PMC7334198
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM,
Ganna A, Birnbaum DP et al.
The mutational constraint spectrum quantified from variation in 141,456 humans.
Nature. 2020 May;581(7809):434-443.
PMID: 32461654; PMC: PMC7334197
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill
AJ, Cummings BB et al.
Analysis of protein-coding genetic variation in 60,706 humans.
Nature. 2016 Aug 18;536(7616):285-91.
PMID: 27535533; PMC: PMC5018207
|
|